
Протео́мика (англ. proteomics) — область молекулярной биологии, посвящённая идентификации и количественному анализу белков (иными словами, высокопроизводительному исследованию белков). Термин «протеомика» был предложен в 1997 году. Совокупность всех белков клетки называют протеомом.
Объектом изучения протеомики являются белки, которые экспрессируются в клетке, ткани или организме в данный момент времени (то есть протеом). Хотя первые методы протеомики, например, секвенирование белков по Эдману, появились задолго до геномных технологий, действительно высокопроизводительное изучение белков стало возможным только в постгеномную эпоху, то есть при наличии известных нуклеотидных последовательностей геномов разных организмов.
Задачи и значение
После геномики и транскриптомики, протеомика — следующий шаг в изучении биологических систем. Основная задача протеомики заключается в идентификации новых белков и их количественном анализе. Соответственно, протеомика объективно сложнее геномики, так как геном организма в большинстве случаев не меняется в ходе жизни, но совокупность всех его белков изменяется постоянно. Различаются даже протеомы клеток разных типов одного организма. Кроме того, изучение протеома осложняется и другими обстоятельствами, например, посттрансляционными модификациями, которым подвергаются многие белки (изучением посттрансляционных модификаций занимаются разделы протеомики — фосфопротеомика и гликопротеомика). Для активности многих белков критически необходимы взаимодействия с другими белками и с РНК, что также осложняет их идентификацию. Наконец, некоторые белки существуют так недолго и столь быстро разрушаются, что их очень сложно зафиксировать имеющимися методами.
Данные, полученные методом протеомики, могут быть использованы для формирования более глубокого понимания причин возникновения разнообразных заболеваний, например, нейродегенеративных, а также разработки методов лечения. С помощью протеомики осуществляется поиск антигенов, пригодных для создания новых вакцин. Идентификация белков, которые аномально экспрессируются при различных раковых заболеваниях, имеет огромное значение для диагностики с помощью биомаркеров, прогнозирования и лечения рака.
Методы
Традиционный подход к изучению белков подразумевает их выделение из тканей и клеток, последующую очистку, в результате чего становится возможным анализировать структуру и функции очищенного белка. Протеомика использует другой подход: всё белковое содержимое клетки можно увидеть и проанализировать в одну стадию. Это стало возможным благодаря появлению и развитию таких методов и технологий, как масс-спектрометрия и двумерный электрофорез. Однако методы протеомики не исчерпываются этими двумя примерами. Ниже рассмотрены использующиеся на данный момент методы исследования белков, в том числе методы количественного анализа и секвенирования аминокислотной последовательности белка, которые на современном этапе используются редко.
Количественный анализ, не требующий информации о структурах белков
Количественный анализ белков с ферментативной активностью можно опосредованно проводить через определение активности этих белков. Ещё в начале XX века подобный анализ можно было осуществить с помощью методов спектрофотометрии. При этом количество катализатора оценивается в условных единицах активности. Условные единицы активности до сих пор используют для описания концентрации в крови таких биомаркеров, как аланинаминотрансфераза и аспартатаминотрансфераза. В 1975 году был разработан способ получения моноклональных антител, и они быстро нашли применение в исследовании белков. Например, если известен антиген данного антитела, то с помощью этого антитела можно идентифицировать исследуемый антиген в экспериментальном образце. В медицине в качестве биомаркеров и в XXI веке широко используются антитела, антигены которых неизвестны, но которые связывают у больных людей гораздо больше антигена, чем у здоровых. Например, гликопротеин CA-125 использовали как биомаркер рака яичников с 1981 года, когда были получены антитела к нему. Значительно позднее идентифицировали сам белок — муцин 16.
Секвенирование последовательности белка
В 1953 году Фредерик Сенгер определил аминокислотную последовательность гормона инсулина. Для мечения и идентификации N-концевого остатка Сенгер предложил использовать 1-фтор-2,4-динитробензол. После связывания с этим реагентом N-концевого остатка белка полипептидную цепь гидролизуют соляной кислотой до отдельных аминокислот и выявляют меченный остаток. Если белок состоит из нескольких полипептидных цепей, то пометятся оба N-концевых остатка, то есть будет установлено число отдельных полипептидных цепей в белке. Для секвенирования всей белковой последовательности чаще применяют метод секвенирования по Эдману.
В 1950-х годах шведский химик Пер Эдман изобрёл метод определения аминокислотной последовательности белков (секвенирование). Первый этап секвенирования по Эдману — обработка исследуемого пептида изотиоцианатом фенила, который взаимодействует с аминогруппой, давая фенилтиокарбомоильный радикал. При умеренном закислении раствора он отщепляется, захватывая вместе с собой N-концевую аминокислоту. В результате в раствор выходит тиазолинон с радикалом, специфичным для данной аминокислоты. Это производное анализируют хроматографически, определяя, какая аминокислота была на N-конце, и цикл повторяется. Если исследуемый белок закреплён на твёрдой подложке, то после каждой обработки изотиоцианатом фенила его можно промывать, удаляя тиазолинон с N-концевой кислотой, и начинать новый цикл. Метод Эдмана позволяет с высокой точностью определять последовательность длиной до 30 аминокислотных остатков. Высокая чувствительность метода также позволяет секвенировать менее 0,1 нмоль пептида с 99 % точностью. Длина полипептидной цепи, которую можно секвенировать методом Эдмана, зависит от эффективности отдельных стадий, которая, в свою очередь, определяется аминокислотным составом полипептида.

В 1960-х годах был создан автоматический секвенатор, реализующий метод Эдмана. Первичную структуру инсулина, на определение которой у Сенгера ушло более 10 лет, в настоящее время можно получить за пару дней прямым секвенированием на белковом секвенаторе. Метод Эдмана сейчас изредка используют при исследовании организмов, геномные последовательности которых неизвестны. Традиционное секвенирование белков также применяют в тех случаях, когда многие их особенности (например, посттрансляционные модификации) нельзя узнать только лишь из последовательности гена.
Большинство белков перед секвенированием необходимо приготовить к нему особым образом. Сначала в белке́ разрушают дисульфидные связи, если они есть, при помощи окисления надмуравьиной кислотой или восстановления дитиотреитолом. Далее белковую цепь дробят на фрагменты протеазами, поскольку секвенирование длинных белков имеет невысокую точность. Обычно для гидролиза используют трипсин, который действует только на те пептидные связи, карбонильная группа которых принадлежит остатку лизина или аргинина. Поэтому, если при полном гидролизе определить число лизиновых и аргининовых остатков в белке, можно предсказать, на сколько фрагментов распадётся белок после обработки трипсином. Полученные фрагменты далее чистят с помощью электрофореза (см. ниже) или хроматографии и секвенируют по Эдману. Чтобы восстановить последовательность белка по фрагментам, его разрезают на куски ферментом, который распознаёт остатки, отличные от тех, которые распознаёт трипсин. На основании перекрытий двух полученных наборов фрагментов восстанавливают полную аминокислотную последовательность белка.
Для определения положения дисульфидных связей белок снова расщепляют трипсином, но не разрушая предварительно дисульфидные связи. Образующиеся фрагменты разделяют электрофорезом и сравнивают с набором фрагментов, полученных при первом расщеплении трипсином. Если между двумя фрагментами есть дисульфидная связь, то при разделении первого набора фрагментов они будут выглядеть на геле как две полосы, а при электрофорезе второго образуют единую полосу.
Двумерный гель-электрофорез

В 1970—1980-х годах достигли расцвета методы выделения и очистки белков. Эти методы сочетали принципы хроматографии, электрофореза и центрифугирования; многие из них давно вышли из употребления, но некоторые используются и в XXI веке. В 1970 году швейцарский учёный Ульрих Лэммли предложил метод разделения белков при помощи электрофореза в денатурирующих условиях. Сначала белки подвергали жёсткой денатурации под действием додецилсульфата натрия (англ. sodium dodecyl sulphate, SDS), который в виде слоя покрывал каждую белковую молекулу. Чем больше был белок, тем больше SDS связывалось с ним и тем больший отрицательный заряд приобретал их комплекс. Поэтому при нанесении образцов на полиакриламидный гель они начинали двигаться под действием электрического поля; при этом скорость движения белковых молекул зависит от их массы (более лёгкие белки перемещаются по гелю быстрее). Метод хорошо подходит для разделения белков с массой от 5 до 250 кДа.

Метод Лэммли получил дальнейшее развитие. В 1975 году Патрик О’Фарелл и Йоахим Клозе независимо друг от друга предложили принцип так называемого двумерного электрофореза: перед разделением по массе с помощью SDS белки предварительно разделяются согласно их изоэлектрической точке. Сначала белки вносят в стеклянную трубку, заполненную особыми полимерами, которые создают в ней неподвижный градиент pH. Белки распределяются по трубке, занимая места, pH которых равен их изоэлектрической точке. Далее содержимое трубки выдавливают и приплавляют к гелю для обычного электрофореза по Лэммли. Таким образом, сначала белки делятся по изоэлектрической точке, а потом по массе. В результате двумерного электрофореза каждому белку соответствует не полоса, как при обычном электрофорезе, а сфокусированное округлое пятно, размер и интенсивность окрашивания которого соответствуют концентрации белка. С помощью двумерного электрофореза можно разделять не только различные белки, но и изоформы одного и того же белка, а также формы белка с разными посттрансляционными модификациями. Были предложены различные усовершенствования методики двумерного электрофореза, некоторые его этапы, а также обработка отсканированных гелей, были автоматизированы. По сути, двумерный электрофорез — единственный способ наглядного представления протеома.
Вестерн-блоттинг

В ряде случаев необходимо установить, с какими клеточными белками взаимодействуют выделенные антитела. Нередко стоит и обратная задача: определить выделенный белок можно с помощью антител, специфически с ним связывающихся. Для этого существует метод вестерн-блоттинга, или иммуноблоттинга. При его применении вначале белки из исследуемого лизата разделяют при помощи гель-электрофореза, а из геля переносят на пористую мембрану. Далее мембрану последовательно обрабатывают антителами, специфичными к искомому белку, и радиоактивно-меченными антителами, связывающимися с первыми антителами. Иногда вместо вторых антител производят ферментативную реакцию с первыми антителами. В результате молекулы искомого белка, распознанные антителами, выявляются как полосы на авторадиограмме или пятна на мембране, по которым можно идентифицировать белок.
Масс-спектрометрия

Масс-спектрометрия включает ряд методов, которые направлены на определение молекулярной массы исследуемых соединений. Она нашла широкое применение и в биологии, в особенности в протеомике. При применении масс-спектрометрии сначала белки, находящиеся в образце, ионизируют, потом в условиях вакуума ионы сортируются и детектируются, давая на выходе спектр, который дальше анализируется специальными вычислительными методами. В конечном итоге для каждого иона определяется значение отношения массы к заряду. Если заряд иона равен единице, то отношение численно равно его молекулярной массе. Поначалу использование масс-спектрометрии в биологии было ограничено из-за того, что ионизация была очень жёсткой и приводила к разрушению молекул. В 1980-х годах был разработан метод ионизации молекул лазером при их сокристаллизации со светочувствительным органическим веществом (его называют матрицей). Матрица окружает молекулы исследуемого вещества и под действием лазера ионизирует соседние молекулы. В некоторых условиях ионизацию можно провести без разрушения исследуемых молекул. Этот метод получил название опосредованная матрицей лазерная десорбция-ионизация (англ. matrix-assisted laser desorption ionization, MALDI). Новый метод ионизации совместили с обычным масс-спектрометрометрическим детектором (времяпролётным, англ. time-of-flight, TOF). В этом детекторе ионы движутся в вакуумной трубке и достигают чувствительной пластины (фотоэлектронного умножителя), которая и является детектором. Время, за которое ион преодолевает длину трубки, обратно пропорционально его массе. В 1990-е и в начале 2000-х годов метод MALDI-TOF очень активно использовался для исследований белков.

Из-за особенностей изотопного разделения пики в спектрах больших белков чрезвычайно сложно анализировать. По этой причине перед исследованием их с помощью фермента трипсина разрушают на пептиды массой 500—2500 Да, и затем по данным для пептидов восстанавливают информацию об исходном белке подобно тому, как при секвенировании нуклеиновых кислот нового поколения исходные последовательности собираются из коротких прочтений. Этот подход называется «протеомикой снизу вверх» (англ. bottom-up). Процесс сборки небезошибочен и приводит к большим потерям информации, поэтому в некоторых случаях исследуются целые белки без расщепления с помощью мощных детекторов сверхвысокого разрешения («протеомика сверху вниз», англ. top-down).
Набор молекулярных масс пептидов, которые были получены при обработке белка трипсином, уникален для каждого белка. Это связано в основном с высокой специфичностью трипсина, который вносит разрез только по остаткам лизина и аргинина. Сравнивая полученную картину молекулярных масс пептидов для исследуемого белка с пептидными картами белков из баз данных, можно установить, какой именно белок исследовался. Этот подход получил название пептидной дактилоскопии. Поскольку полного соответствия экспериментального распределения масс пептидов и эталонных пептидных карт достичь невозможно, была введена количественная оценка (англ. score) вероятности того, что экспериментальная пептидная карта соответствует данной теоретической. Для пептидной дактилоскопии были разработаны специальные программы, например, MOWSE.
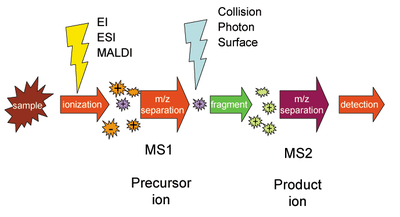
Вместо фрагментации трипсином перед установкой образцов в масс-спектрометр фрагментацию белков на фрагменты можно осуществлять в самом масс-спектрометре, например, при помощи столкновения с молекулами инертных газов. При этом каждый пептид характеризуется массой иона-предшественника и набором масс ионов-фрагментов. Массы фрагментов можно измерить и по ним восстановить информацию об исходном белке, так как молекулярные массы фрагментов можно найти исходя из последовательности пептида. Такой подход получил название тандемной масс-спектрометрии (MS-MS). Как и при пептидной дактилоскопии, в тандемной масс-спектрометрии имеет место вероятностная оценка того, что пептидная карта исследуемого белка соответствует одной из теоретических. В 2007 году для анализа данных тандемной масс-спектрометрии был предложен подход target-decoy. Суть этого подхода заключается в том, что при анализе данных к целевым теоретическим пептидам (англ. target — цель) стали добавлять равное количество бессмысленных, фальшивых (англ. decoy — ложная цель) пептидов. Этот подход позволяет оценить качество анализа. Если анализ в качестве лучших соответствий выдаёт соответствие экспериментального белка с заведомо фальшивым, то он даёт ложноположительный результат, а подход target-decoy позволяет оценить долю ложноположительных результатов.

В качестве альтернативы MALDI ионизацию пептидов перед масс-спектрометрией можно осуществлять с помощью метода ионизации электрораспылением, или ионизации электроспреем (англ. electrospray ionisation, ESI). Жидкость, содержащая исследуемые белки, помещается в конический капилляр, а когда она выходит из капилляра, к ней прилагается сильное напряжение. В результате жидкость превращается в аэрозоль, и при испарении частиц аэрозоля в потоке инертного газа заряд может переходить на растворённые в аэрозоле биомолекулы, в том числе белки. При таком способе ионизации биомолекулы не разрушаются. Ионизацию электроспреем можно легко совместить с высокоэффективной жидкостной хроматографией: поток хроматографической фазы с колонки можно направить прямо в капилляр для электрораспыления. Таким образом, масс-спектрометр будет определять массы разделяемых в аналитической колонке молекул. Этот метод обозначают аббревиатурой LC-MS (от англ. англ. liquid chromatography – mass spectrometry). Идентификация белков в сложном растворе при помощи комбинации масс-спектрометрии и высокоэффективной жидкостной хроматографии получила название протеомики-дробовика, или скорострельной протеомики (англ. shotgun proteomics).
Методы масс-спектрометрии могут быть использованы для направленного обнаружения искомых белков, то есть масс-спектрометр можно настроить таким образом, чтобы он видел только нужный пептид. Для этой цели используют прибор с детектором типа тройного квадруполя, то есть три одинаковых масс-спектрометра, последовательно передающие друг другу ионы. Первый масс-спектрометр отфильтровывает интересующий пептид, во втором он фрагментируется, а третий регистрирует от 3 до 5 заранее выбранных фрагментов. Количественный анализ производится на основе интенсивности фрагментов. Этот метод известен как мониторинг множественных реакций (англ. multiple reaction monitoring, MRM), или мониторинг выбранных реакций (англ. selected reaction monitoring, SRM).
Белок-белковые взаимодействия
Один из наиболее популярных методов изучения белок-белковых взаимодействий — использование дрожжевой двугибридной системы. Для этой цели получают два штамма гаплоидных дрожжей, один из которых исследуемый белок (приманка), а второй — белок, который необходимо проверить на предмет взаимодействия с первым (добыча). Далее гаплоидные клетки сливают с образованием диплоидных клеток дрожжей, экспрессирующих оба белка. Если белки взаимодействуют, то они оба составят транскрипционный фактор, запускающий экспрессию репортёрного гена. Если же взаимодействия между белками нет, то и экспрессия репортёрного гена не запускается. С помощью такого подхода у дрожжей S. cerevisiae при скрининге 6000 клонов добычи против 6000 клонов приманки удалось идентифицировать 691 белок-белковое взаимодействие, из которых только 88 были известны ранее. В XXI веке для исследования белок-белковых взаимодействий применяются и другие методы, такие как плазмонный резонанс.
На основании данных о белок-белковых взаимодействиях в ряде случаев можно судить о функциях белка. Например, если известно, что белок взаимодействует с несколькими белками одного метаболического пути, вполне вероятно, что он тоже в нём задействован. Карты белковых взаимодействий называют интерактом. Существуют базы данных, хранящие информацию о взаимодействиях белков.
Данные о белок-белковых взаимодействий чрезвычайно важны для биологических сетей и системной биологии: они, например, используются при реконструкции сигнальных каскадов.
Белковые микрочипы
Белковые микрочипы разрабатываются для идентификации определённых белков в образце. По аналогии с ДНК-микрочипами, на твёрдую подложку наносятся очень маленькие капли, содержащие антитела. В каждой капле находятся меченые антитела к одному определённому белку, который добавляется на чип в виде флуоресцентно-меченной пробы. После промывки флуоресценция детектируется только в тех каплях, в которых антитела связали исследуемый белок. Вместо антител можно использовать другие молекулы, специфически взаимодействующие с конкретными белками, например, олигонуклеотиды. Белковые микрочипы также можно использовать для обнаружения белок-белковых взаимодействий и определения функций белков. В 2000-е годы белковые микрочипы автоматизированы. Они обладают высокой чувствительностью и требуют совсем небольшого количества исследуемого белка, благодаря чему отличаются экономичностью.
Биоинформатика в протеомике
С помощью масс-спектрометрии и чипов можно получить информацию о фрагментах белка, но не о белке целиком. В связи с этим созданы программы, которые из фрагментарных данных масс-спектрометрии и чипов выдают данные о почти полностью собранных из этих фрагментов белков. Эти программы основаны на построении выравниваний фрагментов с известными белками из баз данных UniProt и PROSITE.
В большинстве программ, анализирующих белки, не учитываются их посттрансляционные модификации. Существующие инструменты, определяющие посттрансляционные модификации, имеют лишь предсказательный характер.
Вычислительные методы биоинформатики активно используются для изучения белков-биомаркеров. Так, с помощью компьютерных моделей удалось показать интенсивный обмен белками между организмом матери и плодом при беременности, причём для анализа требовался лишь неинвазивный забор крови у матери.
Развивается такое направление, как протеогеномика, которая использует методы протеомики для подтверждения данных, полученных из геномных последовательностей. Существует также структурная протеомика, которая занимается широкомасштабным исследованием структур белков на основе данных рентгеноструктурного анализа и ЯМР-спектроскопии.
Протеомика и системная биология
Последние достижения в количественной протеомике позволяют использовать её для глубокого анализа клеточных систем. Описание поведения биологических систем в ответ на разнообразные воздействия (действия внешних факторов, изменения клеточной физиологии в связи с разными фазами клеточного цикла и тому подобные) на уровне изменения белкового состава позволяют глубже понять суть многих биологических процессов. Благодаря этому протеомику, наряду с геномикой, транскриптомикой, эпигеномикой, метаболомикой и другими «-омиками», включают в состав нового научного направления — системной биологии. Так, Атлас протеома раковых клеток (англ. The Cancer Proteome Atlas) содержит количественные данные об экспрессии около 200 белков в более чем 4000 проанализированных опухолевых образцах, дополняя Атлас ракового генома (англ. The Cancer Genome Atlas), содержащий геномные и транскриптомные данные для этих белков.
Практическое применение
С помощью MALDI-TOF можно определять патогенные микроорганизмы с точностью до родов и видов. Интактные бактериальные клетки наносят на металлическую мишень масс-спектрометра, покрывают матрицей, облучают лазером и получают специфичные профили, которые обученный алгоритм распознаёт по характерным массам.
Исследуется возможность использования протеомики для диагностики раковых заболеваний с помощью анализа белковых биомаркеров, а также определения степени злокачественности опухоли. В этом направлении уже достигнуты некоторые успехи. Например, в США разрешено использование разработанного в 2015 году теста Xpresys Lung, который использует таргетную масс-спектрометрию нескольких белков плазмы крови и оценивает степень злокачественности опухолевых узелков в лёгких.
Новейшие достижения протеомики — в области масс-спектрометрии, разделении белков органелл и мембранных белков — могут сделать возможными исследование протеома сердца и идентификацию модифицированных белков (а также определять характер их модификации). Данные по протеому сердца помогут понять механизмы разнообразных сердечно-сосудистых заболеваний.
Многие лекарственные препараты или сами являются белками, или действуют на определённые белки. Поэтому протеомику взяли на вооружение специалисты, занимающиеся разработкой лекарственных препаратов. У большинства фармацевтических компаний есть подразделение, занимающееся протеомикой, или компания-партнёр, специализирующаяся на протеомике. Методы протеомики используют для подтверждения валидности мишеней разрабатываемых препаратов, определения эффективности биомаркеров, изучения механизма действия препарата и его токсичности. Методы протеомики используют, в частности, для поиска противомалярийных препаратов, которые связываются с пурин-связывающими белками на этапе размножения плазмодия в эритроцитах и выхода из них в кровь.
Сравнение протеомов двух организмов (необязательно близкородственных) позволяет выявить как общие для этих двух организмов белки, так и белки, которые обусловливают различия их фенотипов. Такой анализ может давать информацию, полезную для понимания эволюционного процесса, а иногда позволяет определить ранее неизвестные функции белков. Например, при помощи сравнительной протеомики были выявлены белки насекомого Nilaparvata lugens, вовлечённые в процессы, связанные с размножением, чья экспрессия изменяется в ответ на обработку инсектицидами.
История
История протеомики начинается с 1950 года, когда Эдман предложил метод секвенирования белков. В 1958 году исследовательская группа Фредерика Сенгера определила аминокислотную последовательность инсулина. В 1959 году зародился метод иммуноанализа, который имеет огромное значение для изучения белков. В 1967 году был создан первый автоматический секвенатор, определяющий аминокислотные последовательности белков по методу Эдмана. В 1970 году Лэммли предложил метод разделения белков с помощью электрофореза в денатурирующем полиакриламидном геле, а в 1975 году на его основе была предложена методика двумерного электрофореза. В 1984 году был изобретён метод ионизации электроспреем, что позволило изучать белки с помощью масс-спектрометрии без их разрушения, а в 1985 году был предложен метод ионизации MALDI. В 1994 году появились первые пептидные карты для масс-спектрометрии. В 1996 году аспирант Марк Уилкинс ввёл в употребление термин «протеом», и уже в следующем году появился термин «протеомика». В 1999 году появились первые программы для предсказания фрагментов, массы которых будут определены с помощью масс-спектрометрии, по последовательности белка. В 2001 году зародилась скорострельная (англ. shotgun) протеомика, и к 2014 году с помощью этого метода стало возможным идентифицировать 20 тысяч белков человека в одном образце. В настоящее время происходит не только развитие и усовершенствование методов протеомики, таких как различные разновидности масс-спектрометрии, но и новых программ для интерпретации протеомных данных.
Литература
- Принципы и методы биохимии и молекулярной биологии / К. Уилсон и Дж. Уолкер. — М.: БИНОМ. Лаборатория знаний, 2015. — 848 с. — ISBN 978-5-9963-1895-7.
- Нельсон Дэвид, Кокс Майкл. Основы биохимии Ленинджера. В 3 т. — М.: Лаборатория знаний, 2017. — Т. 1. — ISBN 978-5-00101-014-2.