

Mетод присоединения соседей (в лингвистике «метод ближайших соседей») — алгоритм биоинформатики и лингвистики, разработанный Наруя Сайтоу и Масатоcи Нэи в 1987 году. Это восходящий кластерный метод для создания филогенетических деревьев. Обычно используется для деревьев, основанных на ДНК или белковых последовательностях, в лингвистике — на данных лексикостатистики, реже фоно- или морфостатистики. Для его реализации необходимо вычислить расстояния между каждой парой таксонов (например, видов или последовательностей).
Алгоритм

Работа алгоритма начинается с полностью неразрешённого дерева с топологией «звезда».
- Вычисляется матрица попарных расстояний между таксонами.
- По текущей матрице расстояний считается -матрица.
- Ищется пара различных таксонов и (то есть ), для которых значение — наименьшее. Эти таксоны присоединяются к новому узлу, который, в свою очередь, соединяется с центральным узлом. На рисунке справа и присоединены к новому узлу .
- Рассчитывается расстояние от каждого из присоединённых таксонов до нового узла.
- Рассчитывается расстояние от каждого из оставшихся таксонов до нового узла.
- Формируем новую матрицу попарных расстояний: из текущей матрицы удаляем строки и столбцы, соответствующие только что присоединённым таксонам и добавляем новую вершину и, вычисленные в 5 пункте, расстояния.
- Повторяем шаги 2 — 5, пока дерево не станет полностью разрешённым и не станут известны длины всех ветвей.








Q-матрица
-матрица рассчитывается по матрице расстояний между таксонами следующим образом:



где − расстояние между таксонами и .

Расстояние между парой присоединённых соседей и новым узлом
Для каждого из присоединённых таксонов используется следующая формула для расчета расстояний до нового узла:
![{\displaystyle \delta (f,u)={\frac {1}{2}}d(f,g)+{\frac {1}{2(n-2)}}\left[\sum _{k=1}^{n}d(f,k)-\sum _{k=1}^{n}d(g,k)\right]\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/fbb5a4b2a6fbec48a4bbc6d6fc1e351e5eb851e0)

и:

Таксоны и − пара присоединённых таксонов и − новый узел. Ветви и и их длины и − теперь фиксированная часть дерева; они не изменятся и ни на что не повлияют на следующих шагах алгоритма.




Расстояние между оставшимися таксонами и новым узлом
Для каждого таксона, не участвовавшего в предыдущем шаге, рассчитывается расстояние до нового узла:
![{\displaystyle d(u,k)={\frac {1}{2}}[d(f,k)+d(g,k)-d(f,g)]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/29d8e77a0543fb3258d08396a6fb44d387a92ad8)

где — новый узел, — узел, до которого мы хотим рассчитать расстояние, и — таксоны только что присоединённой пары.

Сложность
Метод присоединения соседей для таксонов требует итерации. На каждой итерации требуется рассчитать -матрицу. На первом шаге размер -матрицы — , на следующем шаге — и так далее. Реализация алгоритма без оптимизации имеет сложность ; существуют реализации, использующие эвристический подход с меньшим временем выполнения в среднем.




Пример
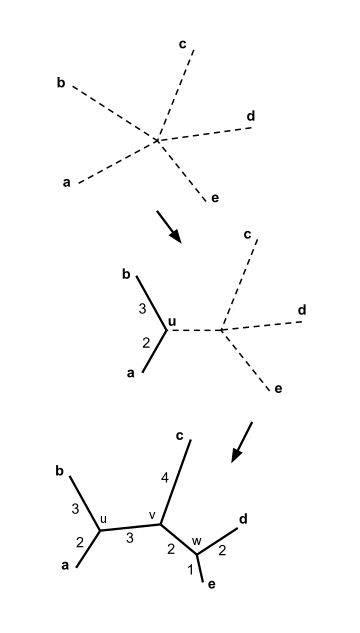
Предположим, у нас есть пять таксонов со следующей матрицей расстояний:

| a | b | c | d | e | |
|---|---|---|---|---|---|
| a | 0 | 5 | 9 | 9 | 8 |
| b | 5 | 0 | 10 | 10 | 9 |
| c | 9 | 10 | 0 | 8 | 7 |
| d | 9 | 10 | 8 | 0 | 3 |
| e | 8 | 9 | 7 | 3 | 0 |
По формуле (1) вычислим -матрицу (диагональные элементы матрицы не используются и здесь опущены):
| a | b | c | d | e | |
|---|---|---|---|---|---|
| a | −50 | −38 | −34 | −34 | |
| b | −50 | −38 | −34 | −34 | |
| c | −38 | −38 | −40 | −40 | |
| d | −34 | −34 | −40 | −48 | |
| e | −34 | −34 | −40 | −48 |
Наименьшее значение матрицы , значит к новому узлу присоединяем таксоны и . Вычислим расстояния от и до по формуле (2):



![{\displaystyle \delta (a,u)={\frac {1}{2}}d(a,b)+{\frac {1}{2(5-2)}}\left[\sum _{k=1}^{5}d(a,k)-\sum _{k=1}^{5}d(b,k)\right]\quad ={\frac {5}{2}}+{\frac {31-34}{6}}=2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/37ab7ad46b3277cb0d8fa097d54007520aadb1df)

По формуле (3) вычисляем расстояния от новой вершины до оставшихся вершин:
![{\displaystyle d(u,c)={\frac {1}{2}}[d(a,c)+d(b,c)-d(a,b)]={\frac {9+10-5}{2}}=7}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7f4eedbd420cf866e0aca938b6b92ac75e7a332d)
![{\displaystyle d(u,d)={\frac {1}{2}}[d(a,d)+d(b,d)-d(a,b)]={\frac {9+10-5}{2}}=7}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4c547916160e8c48cdf86f8c07bf9dc4fb079aa1)
![{\displaystyle d(u,e)={\frac {1}{2}}[d(a,e)+d(b,e)-d(a,b)]={\frac {8+9-5}{2}}=6}](https://wikimedia.org/api/rest_v1/media/math/render/svg/498737ead33eb4e18634803a7c2a940091865cb1)
Таким образом, новая матрица попарных расстояний имеет вид:
| u | c | d | e | |
|---|---|---|---|---|
| u | 0 | 7 | 7 | 6 |
| c | 7 | 0 | 8 | 7 |
| d | 7 | 8 | 0 | 3 |
| e | 6 | 7 | 3 | 0 |
Соответствующая ей -матрица:
| u | c | d | e | |
|---|---|---|---|---|
| u | −28 | −24 | −24 | |
| c | −28 | −24 | −24 | |
| d | −24 | −24 | −28 | |
| e | −24 | −24 | −28 |
Теперь наша матрица принимает минимальное значение на двух парах: , и , . Конечное филогенетическое дерево не зависит от того, какую пару мы выберем. Для определённости, выберем и , присоединим их к новому узлу . Как и в первой итерации вычислим расстояния от и до . Они равны и . Расстояния от новой вершины до оставшихся узлов и равны:






![{\displaystyle d(v,d)={\frac {1}{2}}[d(u,d)+d(c,d)-d(u,c)]={\frac {7+8-7}{2}}=4}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9a149fca7dbf5339b8aaf177e55baf3ae19e3de6)
![{\displaystyle d(v,e)={\frac {1}{2}}[d(u,e)+d(c,e)-d(u,c)]={\frac {6+7-7}{2}}=3}](https://wikimedia.org/api/rest_v1/media/math/render/svg/75942d802678c69bac9b44e8b213994e490fe44c)
Теперь матрица попарных расстояний имеет вид:
| v | d | e | |
|---|---|---|---|
| v | 0 | 4 | 3 |
| d | 4 | 0 | 3 |
| e | 3 | 3 | 0 |
Таким образом, мы получили полностью разрешённое дерево. Однако, для полноты стоит провести ещё одну итерацию:

Матрица попарных расстояний:
| v | d | e | |
|---|---|---|---|
| v | −10 | −10 | |
| d | −10 | −10 | |
| e | −10 | −10 |
Выберем пару и и создадим новую вершину . Расстояния до этой вершины от вершин , , равны соответственно:

![{\displaystyle \delta (v,w)={\frac {1}{2}}d(v,d)+{\frac {1}{2(3-2)}}\left[\sum _{k=1}^{3}d(v,k)-\sum _{k=1}^{3}d(d,k)\right]\quad ={\frac {4}{2}}+{\frac {7-7}{2}}=2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7ef24fb03c3726d7607adbd331c3301b26e40551)


Матрица смежности:
| w | v | d | e | |
|---|---|---|---|---|
| w | 0 | 2 | 2 | 1 |
| v | 2 | 0 | 4 | 3 |
| d | 2 | 4 | 0 | 3 |
| e | 1 | 3 | 3 | 0 |
Таким образом, мы узнали длины всех ветвей и получили полное филогенетическое дерево, показанное на рисунке. Приведённый пример представляет собой идеальный случай: заметим, что если двигаться от одного таксона к другому по ветвям дерева и суммировать длины пройденных ветвей, результат будет равен расстоянию между таксонами в первоначальной матрице расстояний. Например, пройдя от узла к узлу , мы получим . Про матрицу, в которой расстояния согласованы таким образом с каким-либо деревом, говорят, что она аддитивна — свойство, редко встречающееся на практике. Однако важно заметить: если на вход методу присоединения соседей подать аддитивную матрицу расстояний, гарантируется, что в результате работы метода будет построено дерево, согласованное с этой матрицей.

Метод присоединения соседей как минимальная эволюция
Метод присоединения соседей можно рассматривать как жадный алгоритм для оптимизации дерева в соответствии с критерием «сбалансированной минимальной эволюции» (БМЭ). Для каждой топологии БМЭ определяет длину дерева (сумму длин ветвей) как взвешенную сумму расстояний из матрицы расстояний, с весами, зависящими от топологии дерева. Оптимальная топология БМЭ — та, при которой длина дерева минимальна. Метод присоединения соседей на каждой итерации соединяет пару таксонов, которые дадут наименьший вклад в длину строящегося дерева. Эта процедура не гарантирует нахождение дерева с топологией, оптимальной по критерию БМЭ, тем не менее, она часто находит оптимальное или близкое к оптимальному дерево.
Достоинства и недостатки
Главное достоинство метода в том, что он быстрый — в частности, из-за того, что алгоритм работает за полиномиальное время. Это делает его приемлемым для анализа больших объёмов данных (сотни или тысячи таксонов) и для бутстрэпа , для которых использование других методов анализа (например, максимальная экономия, метод максимального правдоподобия) затруднительно с точки зрения количества вычислений.
Метод присоединения соседей имеет свойство выдавать правильное дерево на выходе, если на вход подается правильная матрица расстояний. Более того, правильная топология дерева гарантирована, если матрица расстояний «примерно аддитивна», то есть если каждое значение в матрице расстояний отличается от настоящего расстояния менее, чем на половину длины кратчайшей ветви дерева.
На практике матрица расстояний редко удовлетворяет этому условию, но метод присоединения соседей часто выдает дерево с правильной топологией в любом случае. Метод присоединения соседей корректно работает с примерно аддитивной матрицей расстояний, потому что она статистически состоятельна для многих эволюционных моделей; имея входные данные подходящей длины, метод с высокой вероятностью восстановит настоящее дерево. По сравнению с UPGMA метод присоединения соседей имеет преимущество: он не предполагает, что все поколения эволюционируют с одинаковой скоростью (гипотеза молекулярных часов).
Тем не менее, вместо метода присоединения соседей часто применяют другие филогенетические методы, не полагающиеся на матрицу расстояний и обеспечивающие бо́льшую точность в большинстве случаев.
Реализации и варианты
Существует много программ, реализующих метод присоединения соседей.
RapidNJ и NINJA — быстрые реализации, время работы которых обычно примерно пропорционально квадрату числа таксонов.
BIONJ и Weighbor — варианты метода присоединения, повышающие его точность путём использования факта, что меньшие расстояния в матрице расстояний обычно лучше изучены, чем большие.
FastME — реализация близкого метода сбалансированной минимальной эволюции.
См. также
Ссылки
- The Neighbor-Joining Method — руководство (англ.)