

Анализ взвешенных сетей коэкспрессии генов (англ. weighted gene coexpression network analysis, WGCNA), также известный как анализ взвешенной сети корреляций (англ. weighted correlation network analysis) — метод глубинного анализа данных, основанный на попарных корреляциях между переменными. Метод может быть использован для анализа широкого спектра многомерных наборов данных, но наиболее широкое распространение он получил в геномике. Метод позволяет определять модули (кластеры коэкспрессирующихся генов), межмодульные хабы и узлы сети относительно принадлежности к модулю, изучать отношения между модулями коэкспрессии и сравнивать топологии различных сетей. WGCNA может быть использован как метод снижения размерности данных (связанный с непрямым факторным анализом), как метод кластеризации, как метод отбора признаков (например, для скрининга генов).
История
Метод WGCNA был разработан Стивом Хорватом, профессором Калифорнийского университета в Лос-Анджелесе и членами его лаборатории (в частности, Питером Лангфелдером, Бином Чжаноми и Джуном Донгом) и опубликован в 2005 году. Большая часть метода появилась в ходе прикладных исследований. В частности, взвешенные корреляционные сети были разработаны в рамках совместных дискуссий с исследователями рака Полом Мишелем, Стэнли Ф. Нельсоном, и нейробиологами Дэниэлем Гешвиндом и Майклом Олдхэмом. В 2008 году была опубликована программная реализация алгоритма.
Алгоритм работы
Построение первичной сети
Исходные данные представляют собой матрицу экспрессии генов в нескольких образцах , где — число генов, — число образцов. Для построения сети коэкспрессии для каждой пары генов вычисляется параметр сходства, , который должен лежать в . по умолчанию равен абсолютному значению коэффициента корреляции Пирсона:
![{\displaystyle X_{n\times m}=[x_{il}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/44632ad02d0528985ae4c13c8a7d8813ed6c017f)




![[0;1]](https://wikimedia.org/api/rest_v1/media/math/render/svg/bc3bf59a5da5d8181083b228c8933efbda133483)
,

где профили коэкспрессии генов и состоят из экспрессии генов i и j среди множества образцов. Для достижения большей устойчивости к выбросам можно использовать «урезанный» коэффициент корреляции, а для того, чтобы сохранить знак коэффициента можно использовать простое преобразование корреляции: , так как использование абсолютного значения корреляции может привести к потере биологически значимой информации, поскольку при этом нельзя различить репрессию и активацию генов. Получается матрица сходства .



![{\displaystyle S_{n\times n}=[s_{ij}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/daabca0a7090e63d69b78bcb780c58af90e6a15b)
Фильтрация рёбер по весу с применением мягкого безмасштабного критерия
Для вычисления матрицы смежности сети по матрице сходства необходима функция смежности, которая отображает интервал в интервал . Традиционной функцией смежности является сигнум-функция с жёстким порогом:
![{\displaystyle A_{n\times n}=[a_{ij}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d847c4bfe7f3f9e0e7fc2cfa2f76ca6b885e7a3e)

Такая жёсткая фильтрация рёбер применяется при построении невзвешенных сетей, а результат согласован с интуитивным пониманием концепции сети (связность узла совпадает с числом связанных с ним соседей). Однако такой подход часто приводит к потере информации: например, если установить , то между узлами с параметром сходства 0.79 не будет никакой связи. Таким образом такие сети очень чувствительны к выбору гиперпараметра.

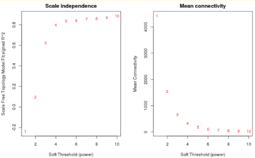
Взвешенные сети лишены этого недостатка. В алгоритме WGCNA в качестве функции смежности используется степенная функция:

Значение определяется с помощью топологического критерия безмасштабности сети для целочисленных значений . Для разных целочисленных значений строится линейная модель зависимости логарифма доли вершин сети со степенью от логарифма степени . Выбирается наименьшее значение , при котором коэффициент детерминации соответствующей линейной модели превосходит 0.8.





«Мягкая» матрица смежности позволяет лишь проранжировать узлы сети согласно силе их связи с рассматриваемым узлом. Если необходимо определить ограниченный список соседей, вводится порог по силе связи. Такой способ «мягкой» фильтрации рёбер сети и называется взвешиванием сети.
Сглаживание взвешенной сети
Для определения модулей сети и удаления шума производится операция сглаживания сети. Сначала вычисляется матрица топологического сходства (topological overlap measure) :
![{\displaystyle \Omega _{n\times n}=[\omega _{ij}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d2511dbfed867aa9a6468e1614dd7298fdcd356c)
,

где — элемент матрицы смежности, , .



равна , если узел с меньшим числом соседей связан с узлом с бо́льшим числом соседей и все соседи узла являются соседями узла . равна , если узлы и не связаны и не имеют общих соседей. Так как , то и . Топологическое сходство двух узлов отражает их относительную внутрисвязанность.







На основе матрицы топологического сходства строится матрица несходства :
![{\displaystyle D_{n\times n}=[d_{ij}^{\omega }]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b69092b2dc9d336bec8ec7d4356194f8944afd99)
.

Модули сети выделяются согласно матрице несходства.

Иерархическая кластеризация и выделение модулей
Для выделения модулей сети используется иерархическая кластеризация матрицы несходства. Модули генов представляют собой ветви полученной дендрограммы. Для выделения модулей производится обрезка ветвей дендрограммы. Статический способ обрезки, который выделяет разветвления ниже определённого порога как отдельный кластер, производит модули, которые легко выделяются визуально, но не соответствуют строгому определению понятия «модуль».
В WGCNA используется метод динамического обрезания дендрограммы (англ. Dynamic Tree Cut), который представляет собой адаптивный итеративный процесс разбиения и комбинации модулей, и останавливается, когда число модулей остаётся стабильным. Исходным набором модулей являются большие модули, определяемые статическим способом. Далее происходит рекурсивное разбиение на подмодули на основе наличия паттерна флуктуаций длины ветвей внутри одного исходного модуля. Маленькие модули объединяются с соседними, чтобы избежать чрезмерного дробления.
Более поздние версии алгоритма используют гибридный динамический (англ. Dynamic Hybrid) подход, который позволяет более успешно детектировать выбросы в каждом кластере. В качестве зачатков модулей используются ветви, удовлетворяющие следующим критериям:
- Содержат не менее установленного порога дочерних листьев (объектов);
- Объекты, расположенные слишком далеко от модуля исключаются из него, даже если они принадлежат одной дочерней ветви на дендрограмме;
- Каждый модуль должен быть различимым от его окружения;
- Ядро модуля должно быть сильно связано.
После определения таких зачаточных модулей все остальные объекты, не попавшие в модулей на первом шаге, по возможности включаются в состав образованных модулей методом ближайших медоидов. Объекты в составе одного итогового модуля могут не находиться рядом на дендрограмме, однако эта несогласованность является обманчивой и следует из ограничений отображения реальных данных в виде дендрограммы.
Валидация модулей
Для валидации модулей используется бутстрэп-анализ на неполных выборках образцов. Дальнейшему анализу подвергаются только те модули, которые были обнаружены в большом числе бутстрэп-реплик.
Интерпретация результатов
Обобщение профиля экспрессии генов модуля
Модуль коэкспрессии представляется собственным геном (англ. eigengene) , который является правым сингулярным вектором, соответствующим наибольшему правому сингулярному значению при сингулярном разложении матрицы экспрессии генов этого модуля .


![{\displaystyle X_{n^{(q)}\times m}^{(q)}=[x_{ij}^{(q)}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0a09338284b8e244750afbf8cb583b18ecb6ed80)

Соотнесение модулей с внешними характеристиками
Для определения значимости влияния экспрессии одного гена на проявление черты можно использовать модуль коэффициента корреляции или p-value соответствующего корреляционного или регрессионного анализа . Для определения значимости влияния экспрессии генов модуля используются разные величины:



- Среднее значение модулей коэффициентов корреляции между экспрессиями генов и проявления черты по данному модулю ;
- Модуль коэффициента корреляции собственного гена и проявления черты и соответствующий p-value регрессионного анализа.



Чем больше величина значимости, тем более значимым является данный ген или модуль генов для проявления данной черты .

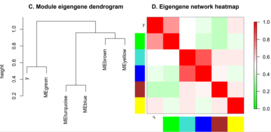
Взаимодействие модулей
Для оценки взаимодействия (коэкспрессии) модулей используются как попарные корреляции между собственными генами модулей, так и построение мета-сети на основе матрицы коэкспрессии собственных генов модулей и выделение мета-модулей в этой мета-сети, в том числе с использованием информации о проявлении черт для определения значимости модулей.
Выделение важных генов
Одним из ключевых моментов в анализе построенной сети является выделение центральных узлов (хабов) — генов, которые высоко скоррелированы со многими другими генами сети. Две метрики связности узла: стандартная и топологическая — могут быть вычислены как по всей сети (полносетевая связность, англ. whole-network connectivity), так и только по генам того же модуля (внутримодальная связность, англ. intramodular connectivity). Стандартная и топологическая полносетевая и внутримодальная связности обозначаются как , , и , соответственно.






Использование внутримодульной связности (нормированной на максимальное значение связности внутри модуля) является более предпочтительным по сравнению с полносетевой связностью при сравнении генов из различных модулей, так как сильно связный ген но из маленького по размеру, но важного модуля может иметь гораздо меньшее значение полносетевой связности по сравнению с геном, имеющим среднюю степень связности из большого по размеру, но не очень важного модуля.
В свою очередь, внутримодульная связность гена отражает то, насколько связан или коэкспрессирован рассматриваемый ген по отношению к генам этого модуля. Эта метрика может быть интерпретирована как мера принадлежности к модулю.
Также выделяют метрики и . Высокое среднее значение для генов модуля (даже превышающее среднее значение ) может означать то, что этот модуль является ядром сети и важным с биологической точки зрения в изучаемых условиях. Высокое значение , напротив, означает, что общая связанность больше, чем связанность внутри модуля, то есть гены, входящие в состав этого модуля, стабильны и слабо меняют экспрессию в исследуемых условиях.



Взвешенная мера принадлежности к модулю
Бинарная мера принадлежности к модулю может быть не самой подходящей мерой для ряда применений, поэтому в качестве меры принадлежности гена к модулю можно использовать непрерывную величину (англ. fuzzy measure of module membership). В качестве такой меры можно использовать или меру связности, основанная на собственных векторах модулей, . Последняя определяется как корреляция гена и собственного гена модуля : . Высокое значение (близкое к или ) говорит о сильной связанности гена и модуля , значение, близкое к , говорит о том, что ген не принадлежит модулю . Метрики и связаны между собой: внутримодальные хабы обладают тенденцией иметь высокое значение для соответствующего модуля.



Сравнение взвешенных и невзвешенных сетей корреляций
Взвешенная корреляционная сеть может рассматриваться как частный случай взвешенной сети, сети зависимостей или корреляционной сети. Анализ взвешенных корреляционных сетей имеет следующие достоинства по сравнению с невзвешенными сетями:
- Построение сети (на основе мягкого порога коэффициента корреляции) сохраняет непрерывный характер исходной информации о корреляции. Например, взвешенные корреляционные сети, построенные на основе корреляций между числовыми переменными не требуют выбора жёсткого порога. Дихотомическое деление информации и (жёсткий) выбор порога может привести к потере информации;
- Взвешенные корреляционные сети облегчают геометрическую интерпретацию на основе угловой интерпретации корреляции;
- Полученная статистика сети может быть использована для улучшения результатов стандартных методов глубокого анализа данных, таких как кластерный анализ, так как меры несхожести зачастую могут быть преобразованы во взвешенные сети;
- WGCNA предоставляет обширные статистические данные о консервативности модулей, которые могут быть использованы для количественного представления возможности встретить их в другом состоянии. Также статистика консервативности модулей позволяет исследовать различия между модульной структурой сетей;
- Взвешенные и корреляционные сети часто можно аппроксимировать факторизуемыми сетями. Таких приближений бывает сложно добиться для разреженных, невзвешенных сетей. Таким образом, взвешенные (корреляционные) сети позволяют использовать экономную параметризацию (в терминах модулей и принадлежности к модулям).
Применение
WGCNA широко применяется для анализа данных об экспрессии генов, например, для поиска межмодульных хабов.
Эта методика часто используется в качестве шага снижения размерности данных в применении к системной генетике, где модули представлены собственными генами. Собственные гены модулей могут быть использованы для расчёта корреляции с экспериментальными данными.
WGCNA широко применяется в нейробиологии и для анализа геномных данных, включая микрочипы, данные RNA-Seq отдельной клетки данные метилирования ДНК,миРНК, подсчёт пептидов и данные микробиоты (секвенирование генов 16S рРНК). Другие области применения включают данные визуализации мозга, например данные функциональной МРТ, анализ онкологических данных, например, выделение подклассов глиом и ассоциированных с ними биомаркеров, а также данные о работе ресничек.
WGCNA можно применять для метаанализа данных, полученных из разных источников.
Программная реализация алгоритма
Пакет программного обеспечения R
Пакет WGCNA программного обеспечения R предоставляет функции для осуществления всех элементов анализа взвешенных сетей (построение модулей, выбор генов хабов, статистика консервативности модулей, дифференциальный анализ сети, статистика сети). Пакет WGCNA доступен в CRAN, стандартном репозитории пакетов дополнений для R, однако для работы WGCNA необходимы пакеты, доступные в репозитории Bioconductor. Пакету посвящён отдельный сайт, где опубликованы краткие руководства с демонстрацией возможностей пакета, а также существует блог одного из авторов метода, в котором публикуются тематические статьи, руководства и новости пакета.
Экспорт результатов для анализа в дочерних программах
Пакет содержит функции для визуализации полученных сетей в VisANT и Cytoscape. Также разработан пакет anRichment для расширенной функциональной аннотации генов в модулях, выделенных WGCNA.
iterativeWGCNA
Существует расширение базового пакета R под названием iterativeWGCNA, реализованное на языке Python. Расширение позволяет усилить устойчивость детектируемых модулей и уменьшить потерю информации. Для этого гены, не попавшие в модули, а также гены со слабым значением связности к своим модулям снова подвергаются WGCNA-анализу и для них заново определяются модули.
Анализ полногеномных данных
Анализ сетей коэкспрессии для наборов данных с большим числом образцов в полногеномном масштабе является вычислительно сложной задачей, требующей большого количества оперативной памяти и времени вычисления. Из-за того, что в процессе построения сети вычисляется коэффициент корреляции между узлами, то анализ сети с узлами требует оперативной памяти и времени вычислений. Существует две стратегии для решения этой проблемы.


Анализ ограниченного набора генов
Можно сократить число анализируемых генов до 4000-5000, которые имеют наибольшее значение какого-либо параметра: значение дисперсии профиля экспрессии генов в массиве данных, значение p-value теста уровня экспрессии генов между различными группами образцов, доля образцов, в которых детектируется экспрессия генов и другие. Основными недостатками такого подхода являются потеря информации об оставшихся генах, систематическая ошибка выборки и неверное распределение генов по функциям.
Эвристические методы определения модулей
Один подход состоит в том, что сначала выделяются модули на некой подвыборке генов, а затем оставшиеся гены добавляются к выделенным модулям на основании метрики : для каждого гена вычисляется, с каким из модулей он сильнее всего коррелирует. Но если данный ген не коррелирует ни с каким модулем выше заданного порога, то он не включается ни в один модуль.
Другой подход состоит в том, что на начальном этапе гены разбиваются на блоки заранее заданного размера с помощью метода k-ближайших соседей, а дальнейшее выделение модулей производится уже внутри этих блоков. Полученные модули сливаются и соотносятся между собой согласно корреляции их собственных генов. Такой подход реализован в пакете WGCNA и для блоков размера требует оперативной памяти и вычислений.



Ссылки
- Steve Horvath. Weighted Network Analysis: Applications in Genomics and Systems Biology. — 1. — 2011. — 414 с. — ISBN 978-1-4419-8818-8.
- Сайт, посвящённый пакету WGCNA
- Обучающие материалы по работе с пакетом WGCNA
- Блог Питера Лангфелдера
- Сайт, посвящённый пакету VisANT
- Сайт, посвящённый пакету anRichment
- Репозиторий пакета iterativeWGCNA