
| Спинальная мышечная атрофия | |
|---|---|
 | |
| МКБ-10 | G12 |
| МКБ-10-КМ | G12.1 и G12.9 |
| МКБ-9 | 335.0-335.1 |
| МКБ-9-КМ | 335.1 и 335.10 |
| OMIM | 253300 |
| DiseasesDB | 14093 |
| MedlinePlus | 000996 |
| MeSH | D009134 |
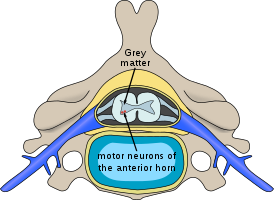
Спина́льная мы́шечная атрофи́я (СМА; англ. spinal muscular atrophy, SMA) — разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга. Генетическое заболевание, при котором возможны все типы наследования: аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный, связанный с мутациями в генах SMN1 и SMN2, кодирующих белок, участвующий в синтезе сплайсосомы.
Для спинальных (их причина находится в спинном мозге) мышечных атрофий характерно нарушение работы поперечнополосатой мускулатуры ног, а также головы и шеи. У больных отмечаются нарушения произвольных движений — ползания младенцев, ходьбы, удержания головы, глотания. Мышцы рук обычно не страдают. Для спинальных амиотрофий характерно сохранение чувствительности, а также отсутствие задержки психического развития.
Общая информация
- СМА — одна из наиболее частых причин детской смертности, вызванной наследственными заболеваниями.
- Спинальные мышечные атрофии детского возраста наследуются по аутосомно-рецессивному типу.
- Ген спинальной мышечной атрофии картирован на 5-й хромосоме, q11.2 — 13.3.
- Ген СМА был идентифицирован в 1995 г., его обозначение SMN (survival motor neuron).
- В среднем один из 6000 — 10000 детей рождается со СМА, в разных странах частота сильно различается.
- 50 % детей с СМА не доживают до двух лет (это дети преимущественно с 1-й формой заболевания).
- SMA может проявиться в любом возрасте, «мягкие» формы проявляются в среднем и пожилом возрасте.
- В среднем каждый 50-й человек имеет рецессивный ген, способный вызывать СМА.
- В соответствии с менделевским расщеплением ребёнок двух носителей поражается СМА с вероятностью 25 %. В этом случае оба родителя несут одиночный дефектный ген, но защищены присутствием второго, нормального гена, который является вообще достаточным для нормальной функции организма. Две дефектных копии гена приводят к генному нарушению, так как не обеспечивается синтез необходимого белка.
- В ходе медико-генетического обследования нескольких российских и среднеазиатских популяций (1,8 млн человек) выявлено 33 больных спинальной мышечной атрофией (СМА): 29 с детской проксимальной СМА (СМА I—III) и 4 c редкими формами. Выявлено «перекрывание» проявлений разных типов СМА I—III (I—II и II—III) у части больных, внутрисемейные различия типов в 3 из 6 семейных случаев, клинико-генетический полиморфизм редких форм СМА. (Г. Е. Руденская, Р. А. Мамедова).
История и патогенез
Спинальная мышечная атрофия у детей впервые была описана Г. Верднигом (G. Werdnig) в 1891 году. Г. Вердниг представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. И. Хофман обосновал нозологическую самостоятельность заболевания. В дальнейшем Вердниг и Хофман (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Э. Кугельберг и Л. Веландер выделили новую нозологическую форму спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной Верднигом и Хофманом.
СМА вызвана мутацией в гене SMN1, который в норме производит белок SMN. Из-за мутации гена у людей с СМА производится меньшее количество белка SMN, что приводит к потере моторных нейронов.
Классификация типов СМА
Выделяют четыре формы проксимальной спинальной амиотрофии на основе возраста начала, тяжести течения и продолжительности жизни.
| Тип | Эпоним | Обычный возраст начала | Описание | OMIM |
|---|---|---|---|---|
| I: Младенческий | СМА I, болезнь Верднига-Хоффмана | 0-6 мес. | Наиболее неблагоприятная форма СМА. Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием, не держат голову, не сидят самостоятельно. | 253300 |
| II: Промежуточный | СМА II, болезнь Дубовица | 7-18 мес. | Больные этой формой спинальной амиотрофии дети могут есть, сидеть, но никогда не достигают способности ходить самостоятельно. Прогноз в этих случаях зависит от степени вовлечения в патологический процесс респираторных мышц. | 253550 |
| III: Юношеский | СМА III, болезнь Кюгельберга-Веландер | >18 мес. | Наименее опасная форма СМА детского возраста. Пациент способен стоять, но испытывает сильную слабость, с тенденцией к инвалидизации (передвижение в коляске). | 253400 |
| IV: Взрослый | СМА IV или взрослая форма | после 35 лет | Значительно не влияет на продолжительность жизни. Слабость проксимальной мускулатуры, фасцикуляции, снижение сухожильных рефлексов. Больные не способны ходить самостоятельно. | 271150 |
Лечение
Радикального лечения не существует.
Так как спинальная мышечная атрофия — нарушение, которое проявляется в синапсах моторных нейронов, состояние может быть улучшено за счёт увеличения уровня SMN — белка. Цель современных исследований — поиск препаратов, увеличивающих уровни SMN. Основные результаты получены пока в исследовательских группах США, Германии, Италии.
Предложено несколько препаратов (вальпроевая кислота, бутират натрия и др.), проводятся их клинические исследования в группах добровольцев. Сведений о результативном применении стволовых клеток пока нет.
Больные СМА нуждаются в специальном диетическом питании, поддерживающей терапии и многих других попечительских действиях, иначе отмечается усиление отрицательной динамики.
В декабре 2016 в США был одобрен первый специализированный препарат для лечения СМА любого типа — «Спинраза» (Spinraza, нусинерсен). Нусинерсен (nusinersen), разработанный «Байоджен» (Biogen) и «Айонис фармасьютикал» (Ionis Pharmaceuticals), представляет собой антисмысловой олигонуклеотид для альтернативного сплайсинга пре-мРНК гена SMN2, который почти идентичен SMN1, и потому синтез полноразмерного белка SMN усиливается. «Спинраза» вводится интратекально.
В мае 2019 года FDA США одобрило «Золгенсма» (Zolgensma, онасемноген абепарвовек) — генную терапию спинальной мышечной атрофии с биаллельной мутацией гена выживаемости моторных нейронов 1 (SMN1) у детей в возрасте младше двух лет. Препарат подходит как симптоматическим пациентам, так и предсимптоматическим, выявляемым генетическим тестированием. Регуляторное разрешение выдано «Авексис» (AveXis), которую «Новартис» (Novartis) купила за 8,7 млрд долларов. Онасемноген абепарвовек (onasemnogene abeparvovec, AVXS-101) представляет собой генотерапевтическое лечение на основе вектора AAV9, после единственной дозы обеспечивающее замену отсутствующего или дефектного гена SMN1 на его функциональную копию без изменения ДНК ребёнка. Итогом становится нормальная выработка белка SMN — и соответствующее прекращение прогрессирования спинальной мышечной атрофии. Во всяком случае однократное введение препарата Золгенсма демонстрирует, что пациенты начинают не только избавляться от зависимости в ИВЛ, но и демонстрировать существенное улучшение моторных навыков (способность сидеть, вставать, ходить), в ряде случаев полностью отвечающих нормальному возрастному развитию. Препарат считается наиболее дорогим лекарством со стоимостью курса (1 укол) более $2 млн. По некоторым оценкам, возможно порядка тысячи применений препарата до 2025 или 2027 года.
«Рош» (Roche) продолжает разработку рисдиплама (risdiplam), который, работая по аналогии с нусинерсеном, характеризуется существенным преимуществом, поскольку сделан в пероральной рецептуре.
Профилактика
Возможна только пассивная профилактика — консультирование родителей с риском СМА о возможных последствиях и пренатальная ДНК-диагностика во время беременности через биопсию ворсин хориона для принятия решения о рождении или прерывании беременности.
СМА в России
Государственный регистр пациентов с диагнозом СМА не ведется. С 2016 года пациентов подсчитывает фонд «Семьи СМА». За это время удалось собрать информацию о 989 пациентах и 250—300 погибших. Теоретически (исходя из общемировой статистики заболеваемости) больных может быть около 2-3 тыс., считают в фонде.
По подсчетам фонда «Семьи СМА», в 2020 году лечением в России обеспечены лишь 129 больных.
В 2020 году единственным зарегистрированным в стране препаратом, показанным для терапии СМА является «Спинраза». Препарат «Спинраза» («Нусинерсен») в России был зарегистрирован в августе 2019 года. Его закупку должны оплачивать региональные власти, однако из-за дороговизны препарата лечение получают не все пациенты. Большинство пациентов, получающих лечение, добились его через суд. В регионах пациенты часто получают отказ в предоставлении лечения. Официально региональные власти объясняют это тем, что заболевание отсутствует в программе ВЗН (высокозатратных нозологий), в перечне жизнеугрожающих орфанных заболеваний или других целевых программах.
В августе 2020 года Миндзрав РФ принял решение о включении препарата в государственный перечень жизненно необходимых и важнейших лекарственных препаратов. Это означает фиксацию предельной отпускной цены на лекарство. Эта мера сделает лечение более доступным.
18 марта 2020 года швейцарская компания Roche («Рош») подала заявку на регистрацию в России препарата рисдиплам (торговое наименование «Эврисди»). В январе компания объявила о запуске глобальной программы дорегистрационного доступа к препарату рисдиплам для пациентов со СМА первого типа. Россия стала первой страной, в которой компания «Рош» открыла возможность участия в программе дорегистрационного доступа к препарату больных со СМА второго типа.
В середине июля 2020 года компания «Новартис» подала досье на регистрацию препарата «Золгенсма» в Министерство здравоохранения РФ. Пока «Золгенсма» не зарегистрирован для клинического применения в России, его ввоз на территорию страны может осуществляться только по жизненным показаниям для конкретных пациентов в соответствии с требованиями, определёнными федеральным законом № 61-Ф3 (об обращении лекарственных средств). Для этого необходимо решение врачебных комиссий медучреждений, а также одобрение на ввоз со стороны Министерства здравоохранения РФ.
Осуществлять применение этого препарата в России могут на сегодняшний день два федеральных центра, которые получили полномочия после прохождения необходимого обучения, организованного производителем «Золгенсма» — компанией АveXis (группа компаний «Новартис»). Оба учреждения являются ведущими российскими медицинскими центрами мирового уровня и имеют богатый опыт работы в педиатрии, включая лечение редких наследственных заболеваний.
На 23 июля 2020 года укол «Золгенсма» получили 16 российских пациентов. До 2021 года терапия «Золгенсмой» государством не финансировалась, все пациенты, прошедшие лечение, получали его за счет средств благотворительных фондов и отдельных меценатов. Известно, что на лечение детей с диагнозом СМА жертвовали средства бизнесмены Владимир Лисин, Владимир Гуриев, Алишер Усманов.
23 июня 2020 года Президент России Владимир Путин выступил с инициативой повышения ставки НДФЛ с 13 % до 15 % для граждан, получающих более 5 млн рублей в год. По предварительным оценкам, эта мера даст бюджету порядка 60 млрд рублей, которые направят на лечение детей с орфанными заболеваниями. Соответствующий законопроект был одобрен Госдумой в первом чтении 21 октября 2020 года. Новый механизм помощи детям с тяжелыми редкими заболеваниями заработал с 1 января 2021 года, когда Президентом был учрежден благотворительный фонд помощи больным детям «Круг добра». К сентябрю 2021 года фонд одобрил финансирование лечения 9 пациентов со СМА «Золгенсмой» из федерального бюджета.
24 ноября 2020 года Правительство России включило препарат «Спинраза» в перечень жизненно необходимых и важнейших лекарственных препаратов. Распоряжение вступило в силу 1 января 2021 года.
Другие формы спинальных мышечных атрофий
Кроме спинальных амиотрофий, обусловленных мутацией в генах SMN1 или SMN2 на длинном плече 5-й хромосомы (5q13.2), вызывающих поражение проксимальных мышц, существует множество схожих заболеваний, большинство из которых — с преимущественным поражением дистальных (то есть ближе к свободному концу конечности) мышц.
| Наименование и синонимы | OMIM | Ген | Локус | Тип наследования | Описание |
| X-сцепленная Спинальная амиотрофия 1-го типа (SMAX1), Spinal and bulbar muscular atrophy (SBMA), Kennedy’s disease (KD) | 313200 | NR3C4 | Xq12 | Х-сцепленный, рецессивный | Позднее начало (в 40-60 лет), медленное прогрессирование, участие в процессе бульбарной группы черепных нервов, нисходящее распространение параличей |
| X-сцепленная Спинальная амиотрофия 2-го типа (SMAX2), Arthrogryposis multiplex congenita — X-linked type 1 (AMCX1) | 301830 | UBA1 | Xp11.23 | Х-сцепленный, рецессивный | Врождённая гипотония и арефлексия вследствие дегенерации и потери двигательных нейронов передних рогов спинного мозга и ствола головного мозга. Часто сочетается с врождёнными контрактурами и/или переломами. Интеллектуальное развитие нормальное. Заболевание быстро прогрессирует, приводя к смерти пациентов до 3-месячного возраста. |
| X-сцепленная Спинальная амиотрофия 3-го типа (SMAX3), Distal spinal muscular atrophy — X-linked (DSMAX) | 300489 | ATP7A | Xq21.1 | Х-сцепленный, рецессивный | Поражены дистальные мышцы всех конечностей, почти всегда у мальчиков, медленно прогрессирующее. |
| Дистальная Спинальная амиотрофия (DSMA1), Spinal muscular atrophy with respiratory distress type 1 (SMARD1), Distal hereditary motor neuropathy type 6 (HMN6) | 604320 | IGHMBP2 | 11q13.3 | Аутосомно-рецессивный | Признаки проявляются с самого рождения, реже во внутриутробном периоде. Заболевание характеризуется преимущественным поражением мышц верхних конечностей и развитием тяжёлых респираторных осложнений из-за прогрессирующей дегенерации мотонейронов передних рогов спинного мозга. |
| Дистальная Спинальная амиотрофия 2-го типа (DSMA2), Distal hereditary motor neuropathy — Jerash type (HMN-J) | 605726 | ? | 9p21.1-p12 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье |
| Дистальная Спинальная амиотрофия 3-го типа (DSMA3), Distal hereditary motor neuropathy types 3 & 4 (HMN3, HMN4) | 607088 | ? | 11q13.3 | Аутосомно-рецессивный | Медленно прогрессирующее |
| Дистальная Спинальная амиотрофия 4-го типа (DSMA4) | 611067 | PLEKHG5 | 1p36.31 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье |
| Дистальная Спинальная амиотрофия 5-го типа (DSMA5) | 614881 | DNAJB2 | 2q35 | Аутосомно-рецессивный | Начинается в молодом взрослом возрасте, медленно прогрессирующее. |
| Дистальная Спинальная амиотрофия VA-типа (DSMAVA), Distal hereditary motor neuropathy type 5A (HMN5A) | 600794 | GARS | 7p14.3 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. |
| Дистальная Спинальная амиотрофия VB-типа (DSMAVB), Distal hereditary motor neuropathy type 5B (HMN5B) | 614751 | REEP1 | 2p11 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. |
| Дистальная Спинальная амиотрофия с преимущественным поражением голеней, Distal hereditary motor neuropathy type 2D (HMN2D) | 615575 | FBXO38 | 5q32 | Аутосомно-доминантный | Проявляется в юношестве или у взрослых, медленно прогрессирует, поражает проксимальные и дистальные мышцы, сначала проявляется слабость в голенях, которая распространяется и на руки. |
| Дистальная спинальная амиотрофия с преимущественным поражением голосовых связок, Distal hereditary motor neuropathy type 7A (HMN7A), Harper-Young myopathy. | 158580 | SLC5A7 | 2q12.3 | Аутосомно-доминантный | Проявляется у взрослых параличом голосовых связок, очень редкое заболевание. |
| Аутосомно-доминантная Спинальная амиотрофия, Distal hereditary motor neuropathy type 2A (HMN2A) | 158590 | HSPB8 | 12q24.23 | Аутосомно-доминантный | Проявляется у взрослых. Аллельный вариант болезни Шарко-Мари-Тутса (CMT2L) |
| Аутосомно-доминантная ювенильная Спинальная амиотрофия, Distal hereditary motor neuropathy type 1 (HMN1) | 182960 | ? | 7q34-q36 | Аутосомно-доминантный | Проявляется в юном возрасте |
| Врождённая дистальная Спинальная амиотрофия | 600175 | TRPV4 | 12q24.11 | Аутосомно-доминантный | Поражение двигательных нейронов спинного мозга, иннервирующих нижнюю часть тела. Проявляется непрогрессирующей мышечной атрофией, атрофией мышц бёдер, мышц-разгибателей стопы, слабостью в коленях. Формируются контрактуры в коленных суставах и деформируются стопы. У некоторых пациентов может наблюдаться паралич голосовых связок. |
| Лопаточно-малоберцовая Спинальная амиотрофия (SPSMA), Scapuloperoneal neurogenic amyotrophy | 181405 | TRPV4 | 12q24.11 | Аутосомно-доминантный или Х-сцепленный, доминантный | Поражает мышцы нижних конечностей. Очень редкое заболевание. Аллельный вариант Врождённой дистальная Спинальной амиотрофии. |
| Ювенильная сегментальная Спинальная амиотрофия (JSSMA) | 183020 | ? | 18q21.3 | ? | Начинается в юности, прогрессирует 2-4 года, после чего стабилизируется, влияет в первую очередь на руки, очень редкое. |
| Спинальная амиотрофия Финкеля, Finkel-type proximal spinal muscular atrophy (SMA-FK) | 182980 | VAPB | 20q13.32 | Аутосомно-доминантный | Средний возраст манифестации заболевания 37 лет (известны случаи в возрасте до 12 лет). Симметричная мышечная слабость и истощение мышц. Медленная потеря мышечной силы и прогрессирующая проксимальная атрофия, которая начинается в ногах и со временем распространяется на руки. Также у больных наблюдаются генерализованные фасцикуляции, гипоактивность или отсутствие глубоких сухожильных рефлексов. |
| Спинальная амиотрофия Джокела, Jokela-type spinal muscular atrophy (SMA-J) | 615048 | ? | 22q11.2-q13.2 | Аутосомно-доминантный | Позднее начало, медленно прогрессирование, поражает проксимальные и дистальные мышцы у взрослых. |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 1, Spinal muscular atrophy with lower extremity predominance 1 (SMALED1) | 158600 | DYNC1H1 | 14q32 | Аутосомно-доминантный | Поражает проксимальные мышцы у младенцев. |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 2, Spinal muscular atrophy with lower extremity predominance 2 (SMALED2) | 615290 | BICD2 | 9q22.31 | Аутосомно-доминантный | Врождённое или с ранним началом, поражающее преимущественно нижние конечности, не прогрессирует, очень редкое. |
| Спинальная амиотрофия с прогрессирующей миоклонической эпилепсией, Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME), Jankovic-Rivera syndrome. | 159950 | ASAH1 | 8p22 | Аутосомно-рецессивный | Медленно прогрессирует, преимущественно поражает дистальные мышцы, сочетается с денервацией и миоклоническими приступами. |
| Спинальная амиотрофия с атрофия с врожденными переломами костей, Spinal muscular atrophy with congenital bone fractures (SMA-CBF) | 271225 | ? | ? | Аутосомно-рецессивный? | Тяжёлое истощение мышц (как при болезни Верднига-Гоффмана), сопровождается врожденными переломами костей. |
| Спинальная амиотрофия с понтоцеребеллярной гипоплазией, Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH), Pontocerebellar hypoplasia type 1A (PCH1A) | 607596 | VRK1 | 14q32 | Аутосомно-доминантный | Описано восемь типов понтоцеребеллярной гипоплазии. Частота заболеваний неизвестна. Все формы заболевания имеют общие признаки: аномальное развитие головного мозга, проблемы с двигательной активностью, задержку развития, умственную неполноценность, прогрессирующую микроцефалию и церебральные проявления различной степени. Заболевание проявляется с рождения, в ряде случаев первые признаки отмечаются уже внутриутробно. Пациенты, как правило, погибают в раннем детском возрасте. |
| Ювенильная асимметричная сегментальная Спинальная амиотрофия, Juvenile asymmetric segmental spinal muscular atrophy (JASSMA), Monomelic amyotrophy; Hirayama disease; Sobue disease | 602440 | ? | ? | ? | Болезнь мотонейронов, которая поражает молодых (15-25-летних) мужчин в Индии и Японии. Начинается с мышечной атрофии, которая стабилизируется в плато после 2-5 лет, симптоматика не меняется. Нет боли или потери чувствительности. В отличие от других более низких мотонейронных болезней, MMA, как полагают, не наследуется и редко проявляется фасцикуляциями. |
Ссылки
- Отдел по подготовке родителей детей с СМА, Милан, Италия
- Украинский Фонд для помощи пациентам с СМА, украинский реестр
- форум по СМА
- Российский Благотворительный фонд «Семьи СМА»
- Европейская сеть нервномышечных заболеваний (СМА, DMD, международный регистр)
- SMA Europe
- (англ. Фонд Великобритании) — Jennifer Trust
- (англ. Фонд Великобритании) — The SMA Trust
- (недоступная ссылка) (англ. Фонд США) — Families of Spinal Muscular Atrophy (недоступная ссылка)
- (англ. Фонд США) — Fight Spinal Muscular Atrophy
- Статья о конверсии между генами при СМА, северо-западный регион России
- Лопаточно-бедренная спинальная мышечная атрофия // Неврологический журнал, № 2, 1998)
- «Золдженсма»: генная терапия, которая вылечит спинальную мышечную атрофию. Все подробности
- Рисдиплам: год лечения спинальной мышечной атрофии
См. также
- Боковой амиотрофический склероз (OMIM 105400)
- Спастическая спинальная параплегия
- Детский церебральный паралич
Литература
- А.Н. Бакланов, С.В. Колесов, И.А. Шавырин. Хирургическое лечение тяжелых нейромышечных сколиозов у пациентов, страдающих спинальной мышечной атрофией // Хирургия позвоночника. — 2011. — № 3. — С. 31—37. — ISSN 1810-8997.