
Выра́внивание после́довательностей — биоинформатический метод, основанный на размещении двух или более последовательностей мономеров ДНК, РНК или белков друг под другом таким образом, чтобы легко увидеть сходные участки в этих последовательностях. Сходство первичных структур двух молекул может отражать их функциональные, структурные или эволюционные взаимосвязи. Выровненные последовательности оснований нуклеотидов или аминокислот обычно представляются в виде строк матрицы. Добавляются разрывы между основаниями таким образом, чтобы одинаковые или похожие элементы были расположены в следующих друг за другом столбцах матрицы.
Алгоритмы выравнивания последовательностей также используются в NLP.
Графическое и текстовое представление
В большинстве представлений результата выравнивания последовательности располагаются в строчках матрицы таким образом, что совпадающие элементы (нуклеотиды или аминокислоты) находятся один под другим (в одной колонке). «Разрывы» заменяются знаком «-», именуемый гэпом (от англ. «gap»), и обозначает индель, то есть место возможной вставки или делеции.
Текстовое представление
При текстовом отображении возможна просто запись в формате fasta, когда последовательности записываются с гэпами, и имеют одинаковую длину. Такой вид записи часто используется программами, и удобен для машинной обработки.
Другой вид текстового представления служит для удобства пользователя (три разных примера представлены ниже). В нём последовательности записываются одна под другой, а в строчке между ними разными символами обозначены разные отношения между аминокислотами. Пробелом (отсутствием символа) обозначают отсутствие связи между аминокислотами, как по гомологии, так и по функции: символами «*», "|" или же буквой (BLAST) — одинаковые аминокислоты; «:» или «+» — близкие по свойствам; «.» — сходные по свойствам.
Blast:
Query 15 FQQAWANPKHAWAQVNGETRLTQNLIILERETR 47
F W PKHA +QVNG T ++Q+ IIL R R
Sbjct 14 FHHNWTRPKHASSQVNGHTEMSQHNIILRRVPR 46
CLUSTAL:
THE12851.1 MGKKGYKRNEYNNPFQQAWANPKHAWAQVNGETRLTQNLIILERETRKRS- 50
WP_104057486.1 MSTK-DQLDPQSQAFHHNWTRPKHASSQVNGHTEMSQHNIILRRVPRSGRR 50
*..* : : .: *:: *:.**** :****.*.::*: ***.* *.
EMBOSS Needle:
THE12851.1 1 MGKKGYKRNEYNNPFQQAWANPKHAWAQVNGETRLTQNLIILERETRKRS 50
|..|. :.:..:..|...|..||||.:||||.|.::|:.|||.|..|...
WP_104057486. 1 MSTKD-QLDPQSQAFHHNWTRPKHASSQVNGHTEMSQHNIILRRVPRSGR 49
Графическое представление
Графическое представление максимально ориентированно на визуальное восприятие. В нём также принято размещать последовательности одну под другой, но значение связи между аминокислотами из разных последовательностей обозначаются цветом. Есть окраски по свойствам аминокислот, такие как «Zappo», окрашивающая каждую аминокислоту, и «Clustal», окрашивающая столбцы с одинаковыми свойствами аминокислот. Часть окрасок, такие как «%Identity», позволяет увидеть идентичность и консервативность аминокислот в столбце. Есть и окраски, показывающие степень гидрофобности аминокислот.
Наиболее известные программы для просмотра выравниваний: Jalview, UGENE, MEGA. Полный список представлен в статье «List of alignment visualization software» (англ).
Также существует способ представления консенсусной последовательности — Логотип последовательности.
Точечная матрица
Точечная матрица сходства — способ визуального представления парного выравнивания. Обычно используется для больших последовательностей, например для геномов бактерий. По осям отложены координаты обеих последовательностей, а отрезками отображают их гомологию. Так, точечная матрица одинаковых последовательностей будет выглядеть как диагональ квадрата. Такой способ представления позволяет отслеживать инверсии, дупликации или делеции, а также транслокации.
Парное выравнивание
Парное выравнивание используется для нахождения сходных участков двух последовательностей. Различают глобальное и локальное выравнивание. Глобальное выравнивание предполагает, что последовательности гомологичны по всей длине. В глобальное выравнивание включаются обе входные последовательности целиком. Локальное выравнивание применяется, если последовательности содержат как родственные (гомологичные), так и неродственные участки. Результатом локального выравнивания является выбор участка в каждой из последовательностей и выравнивание между этими участками.
Для получения парного выравнивания используются разновидности метода динамического программирования. В частности, эти алгоритмы реализованы в сервисах европейской молекулярно-биологической лаборатории (Pairwise Sequence Alignment. EMBL-EBI.). Так, например, Needle., алгоритм глобального выравнивания, использует алгоритм Нидлмана — Вунша, а Water., алгоритм локального выравнивания — алгоритм Смита — Ватермана.
Сравнение глобального и локального выравниваний
Для демонстрации в чём отличие глобального и локального выравниваний, можно рассмотреть искусственный пример. Возьмём последовательности A и B, и сделаем для них глобальное и локальное выравнивание. В последовательности был заложен центральный гомологичный участок, и заметно отличающиеся края.

Глобальное выравнивание использует полную длину обеих последовательностей, и может быть использовано для проверки последовательностей на гомологию (общность происхождения) по всей длине. Однако, если последовательности имеют мало участков гомологии (или просто схожести), то не всегда можно хорошо определить эти участки. В приведённом примере алгоритм зацепился за четыре совпадающий аминокислоты, так что длинный участок гомологии не виден. На основании этого можно предположить, что последовательности целиком не гомологичны между собой.
Локальное выравнивание использует части последовательностей, на которых прогнозируется максимальная гомология. Оно отлично подходит, если лишь части последовательностей похожи, например в ходе рекомбинации или конвергентной эволюции. Всегда стоит аккуратно относиться к небольшим участкам имеющим низкое сходство, особенно при выравнивании больших последовательностей, так как повышается вероятность встречи случайного схожего участка. В примере на рисунке локальное выравнивание включило половину длины последовательностей. Выравнено 11 аминокислот сходных по функции, имеется 2 инделя. На основании этого, если дополнительно известно о схожей функции пептидов A и B, можно сказать, что центральные участки обоих пептидов выполняет функцию всего пептида, либо же важны для его функции.
Однако, не всегда в локальное выравнивание может попасть интересующий участок последовательности. Это можно обойти, если обрезать последовательность по границам интересующего участка. Также возможны и другие комбинации глобального и локального выравниваний.
Алгоритмы поиска
Применяются для поиска в больших базах данных последовательностей, схожих с некой заданной последовательностью по указанным критериям. Применяемое выравнивание — локальное. Для повышения скорости поиска используются различные эвристические методы. Наиболее известные программы: BLAST и FASTA3x..
Множественное выравнивание
Множественное выравнивание — это выравнивание трёх и более последовательностей. Применяется для нахождения консервативных участков в наборе гомологичных последовательностей. В большинстве случаев построение множественного выравнивания — необходимый этап реконструкции филогенетических деревьев. Нахождение оптимального множественного выравнивания методом динамического программирования имеет слишком большую временную сложность, поэтому множественные выравнивания строятся на базе различных эвристик. Наиболее известные программы, осуществляющие множественное выравнивание — Clustal (clustal.), T-COFFEE (tcoffee.), MUSCLE (muscle.) и MAFFT (mafft.). Имеются также программы для просмотра и редактирования множественных выравниваний, например Jalview или русскоязычный UGENE.
Структурное выравнивание
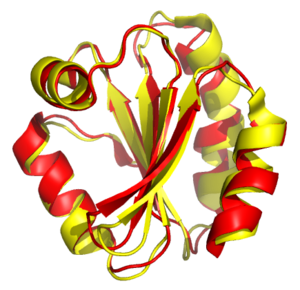
Может быть построено для белков или рибонуклеиновых кислот с использованием информации о вторичной и третичной пространственной структуре молекул. Целью является попытка установить гомологию двух или нескольких структур путём нахождения и сопоставления участков, одинаково уложенных в пространстве. Структурное выравнивание обычно сопровождается наложением структур, то есть нахождением движений пространства, применение которых к заданным молекулам наилучшим образом совмещает их. Но в отличие от простой пространственной суперпозиции с известным сопоставлением эквивалентных аминокислотных остатков двух структур, алгоритмы структурного выравнивания обычно не требуют априорного знания выравнивания последовательностей. Существует большое количество алгоритмов, на которых основаны различные программы структурного выравнивания. Пространственные выравнивания особенно важны для анализа данных структурной геномики и протеомики, они также могут использоваться для оценки выравниваний, полученных путём сравнения последовательностей..
Структурное выравнивание успешно используется для сравнения белков с низким уровнем гомологии последовательностей, когда эволюционные связи не могут быть установлены стандартными методами выравнивания последовательностей, но в этом случае необходимо принимать во внимание влияние конвергентной эволюции, основной эффект которой проявляется в сходстве третичных структур неродственных аминокислотных последовательностей.
Пространственное выравнивание позволяет сравнивать две и более молекулы с известными трёхмерными структурами, экспериментальное получение которых основано на использовании методов рентгеноструктурного анализа и ЯМР-спектроскопии. Для пространственного выравнивания можно также использовать структуры, полученные методами предсказания структуры белка. Более того, оценка качества таких предсказаний зачастую базируется на использовании пространственного выравнивания структуры создаваемой модели и белка, третичная структура которого получена непосредственно из эксперимента. Также есть данные об использовании метода малоуглового рентгеновского рассеяния для анализа трёхмерных структур различных белковых молекул.
Типы сравнений
Результатом работы программ структурного выравнивания, как правило, является совмещение наборов координат атомов. Чаще всего при поиске такого сопоставления оценка результату даётся исходя из значения функции наименьшего среднеквадратического отклонения (RMSD) между структурами, которое алгоритм построения выравнивания старается минимизировать.
- ,

где — количество точек (атомов) в выборке (структуре), и — атомы соответствующей структуры, имеющие координаты , , и , , .









Значение RMSD выражается в единицах длины, наиболее часто используемой единицей в структурной биологии является Ангстрем (Å), который равен 10−10 м. Однако RMSD как степень пространственного расхождения выравниваемых структур имеет ряд недостатков: неустойчивость к выбросам и наличию нескольких доменов в структуре выравниваемых белков, так как изменения в относительном расположении этих доменов между двумя структурами могут искусственно изменять значение RMSD.
Кроме того, могут быть рассчитаны и более сложные параметры, оценивающие структурное сходство, например, тест глобальных расстояний.
Для создания структурного выравнивания и подсчёта соответствующих значений RMSD могут быть использованы как все атомы, входящие в молекулу белка, так и их подмножества. Например, атомы боковых радикалов аминокислотных остатков учитываются не всегда, и для выравнивания могут использоваться только атомы, входящие в пептидный остов молекулы. Такой вариант выбирают, если у выравниваемых структур очень разная аминокислотная последовательность и боковые радикалы различаются у большого числа остатков. По этой причине по умолчанию методы пространственного выравнивания используют только атомы остова, вовлечённые в пептидную связь. Для большего упрощения и увеличения эффективности часто используется положение только альфа-атомов углерода, так как их положение достаточно точно определяет положение атомов полипептидного остова. Только при выравнивании очень похожих или даже идентичных структур важно учитывать позиции атомов боковых цепей. В этом случае RMSD отражает не только схожесть конформации белкового остова, но и ротамерные состояния боковых цепей. Другие способы, позволяющие снизить шум и увеличить число правильных сопоставлений, используют разметку элементов вторичной структуры, карты нативных контактов или паттерны взаимодействия остатков, меры степени упаковки боковых цепей и меры сохранения водородных связей.
Методы
DALI
Одним из популярных методов структурного выравнивания является DALI (англ. distance alignment matrix method — метод с использованием матрицы дистанционных выравниваний). Исходные структуры белков разбиваются на гексапептиды и через оценку паттернов контактов между фрагментами рассчитывается матрица расстояний. Элементы вторичной структуры, остатки которых являются соседними в последовательности, оказываются на главной диагонали матрицы; остальные диагонали матрицы отражают пространственные контакты между остатками, которые в последовательности не находятся рядом друг с другом. Когда матрицы расстояний двух белков имеют одинаковые или похожие элементы примерно на одинаковых позициях, можно сказать, что белки имеют схожую укладку и их элементы вторичной структуры соединены петлями примерно одинаковой длины. Непосредственный процесс выравнивания DALI заключается в поиске схожестей матриц, построенных для двух белков, которые потом пересобираются в конечное выравнивание с помощью стандартного алгоритма максимизации счёта.
Метод DALI был использован для создания базы данных FSSP (англ. Families of Structurally Similar Proteins), в которой все известные структуры белков были попарно выровнены для определения их пространственного родства и классификации укладок.
DaliLite является скачиваемой программой, использующей алгоритм DALI.
Комбинаторное расширение (combinatorial extension)
Метод комбинаторного расширения (англ. Combinational extension (СЕ)) похож на DALI тем, что тоже разбивает каждую структуру на ряд фрагментов, которые затем пытается заново собрать в полное выравнивание. Серия попарных сочетаний фрагментов, называемых AFP (англ. aligned fragment pairs — пары выровненных фрагментов), используется для задания матрицы сходства, через которую прокладывается оптимальный путь для определения конечного выравнивания. Путь, соответствующий выравниванию, рассчитывается как оптимальный путь через матрицу сходства с помощью линейного прохода через последовательности, расширяя выравнивание следующей возможной AFP с высоким счётом. Только те AFP, которые удовлетворяют заданным критериям локального сходства, включаются в матрицу, что сокращает необходимое пространство поиска и увеличивает эффективность.
Подобно DALI или SSAP, CE использовался для создания базы данных классификации укладок на основе известных пространственных структур белков из PDB.